
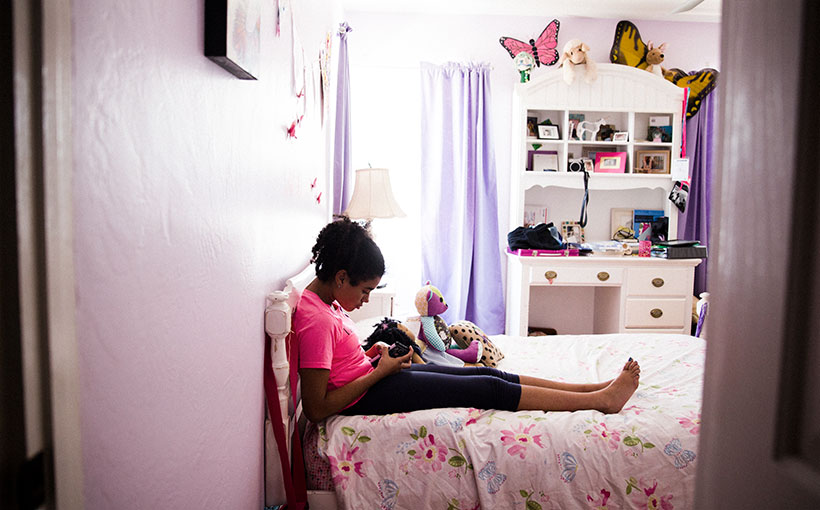
Maya James is in her sixth-grade classroom waiting for her mom to pick her up. She sits quietly, smiling to herself. Perhaps she is thinking about the school day or what she will do when she gets home. She has that glow of boundless possibilities.
But as Maya stands, she seems to rise and crumple at the same time. Unable to walk on her own, she leans heavily on her mother as they move toward the parking lot.
Maya has a rare neurodegenerative condition—a form of Batten disease called atypical TPP1 deficiency. The word “rare” hardly does it justice: ultra rare, hyper rare, twenty-people-in-the-entire-world rare?
Atypical TPP1 deficiency is a lysosomal storage disorder. Lysosomes are like cellular garbage trucks, and sometimes they go on strike. Waste doesn’t get collected; cells get sick and die. When working properly, TPP1 grabs old and damaged proteins and recycles them into their component amino acids.
“It takes everything.”
It’s a double whammy for Maya. The accumulated garbage interferes with cellular function, and without the recycled aminos, cells have trouble making new proteins. The neurological aspect is even worse. Dead neurons rarely get replaced.
Her parents, Beau and Suzette, are on it, but there’s no cure and virtually no treatments. The disease manifests as ataxia—the loss of coordinated muscle movement. If no therapies are found, Maya will gradually lose the ability to walk, speak, eat, and ultimately, live.
“It’s everything,” says Suzette. “It takes everything.”
On an Island
By definition, a rare disease affects fewer than 200,000 people in the United States. But there are thousands of these conditions, affecting around 30 million Americans altogether.
Simply getting information can be daunting. Most physicians have never seen, let alone heard of, any given rare disease.
“It’s a difficult position for the physician providing that diagnosis,” says Jennifer Friedman, Maya’s neurologist at Rady Children’s Hospital. “I will learn what I can and help direct the family, but at some point the baton gets handed over. The parents have to become the experts.”
The Jameses have not shrunk from the challenge. Suzette is a former management consultant and Beau is an attorney at Qualcomm. Their collective drive can be measured in gigawatts, and they’ve needed every bit.
The two have immersed themselves in the opaque world of medical research: reading papers, attending conferences, contacting researchers, hunting for the medical guidance they would normally get from physicians.
“My chemistry and bio teachers are somewhere laughing,” says Beau. “I was the kid in the back saying, ‘I’ll never use this.’”
Without sequencing, they might still be waiting.
They spent a horrific year not knowing what was causing Maya’s decline. Eventually, clinicians at Baylor University Medical Center sequenced her exome—the small percentage of DNA that codes for proteins—and found the TPP1 anomaly. Without sequencing, they might still be waiting.
But they’re living in a therapeutic no-man’s land. Like many rare disease families, they have a diagnosis but no treatments. The Orphan Drug Act—the 1983 law that provides economic incentives to help companies address some of these conditions—has led to new treatments, but progress is slow. There are more than 7,000 rare diseases and fewer than 500 approved therapies for them.
Families around the world band together on Facebook, share blog posts, or visit rare disease websites like Global Genes to find support. But ultimately, the burden rests on parents.
“If you had a cancer diagnosis, you would have a case manager calling you the next day,” says Suzette. “You would be walked through the steps; you’d be given support group meetings. There’s none of that with a rare disease. You’re pretty much on an island, on your own, by yourself.”
The task list can be overwhelming. Parents may be forced to advocate for accommodations at school, lobby drug companies to get their child into a clinical trial, or even start a nonprofit. John and Aileen Crowley started a biotech company to help their children with Pompe disease, a story made famous in the movie Extraordinary Measures.
Tracy Dixon-Salazar’s daughter, Savannah, developed a rare form of epilepsy called Lennox-Gastaut syndrome, sometimes seizing five or more times a day. Dixon-Salazar went back to school to better understand the biology, eventually earning her PhD.
While conducting her postdoctoral research at UC San Diego, she had Savannah’s exome sequenced and personally sifted through her daughter’s genetic variations. Ten months later, she found the culprit—a series of mutations in calcium channel genes, which govern a cell’s electrical activity. More than 16 years after Savannah’s first attack, an existing heart medication reduced her seizures manifold.
That’s the hope that drives rare disease parents. The answers are there, waiting to be teased out. But time is not on their side.
“You have to be resourceful, but you’re drowning,” says Beau. “There’s so much you have to do, and you’re just trying to get your kid through the day.”
A Therapeutic Guide
There’s only so much DIY medical care a family can provide. They need a therapeutic expert, a Virgil to guide them through their personal hell. In some cases, it’s a medical researcher.
“Parents who find their child has one of these disorders really have no place to turn,” says Hudson Freeze, who directs the Human Genetics Program at Sanford Burnham Prebys Medical Discovery Institute. “Their physicians have gone to the limit of what they know, so they have to turn to the research community, probably not something they ever thought they’d have to do.”
Freeze has spent decades working on congenital disorders of glycosylation. Many proteins only work properly if attached to sugars, and when that process goes awry, bad things happen. Freeze has helped dozens of families, providing invaluable information about the aberrant molecules that are causing their kids’ conditions. Pictures of children decorate his lab.
“You have to admire these parents,” he says. “They go to the ends of the earth to find out about their child’s disease.”
Beau and Suzette have scoured the research community to find therapeutic guides. Kalipada Pahan, a professor of neurological sciences at Rush University, suggested they try vitamin A and gemfibrozil, supplements that could help Maya hold on to neurons. She also takes turmeric and eats lots of fruits and veggies—anything to reduce the inflammation associated with her disease.
“We started with Pahan and his research, and he was able to increase the enzyme two or three levels,” says Beau. “Now, who can get me four to eight more? We’re going to stair-step it until we get something really good.”
Racing for a Treatment
Though it’s hardly any comfort, Maya has been lucky. Ten years ago, there would have been no sequencing to provide a diagnosis.
“We are in a very different situation from many families,” says Suzette. “There are kids that don’t have a diagnosis, and they certainly don’t have any options for treatment.”
In addition, her form of Batten disease is less aggressive than a more common variant, called CLN2, which strikes earlier and is even more deadly.
BioMarin, a Bay Area biotech company, is developing a CLN2 drug that is now in clinical trials.
Spark Therapeutics is working on gene therapy, but the treatment is still in the lab and won’t go to the clinic for years.
Maya’s best hope may be Marco Sardiello, an assistant professor of Molecular and Human Genetics at the Jan and Dan Duncan Neurological Research Institute at the Baylor College of Medicine. Sardiello’s lab is working on a protein called TFEB, a “transcription factor” or master switch that turns on other genes. In this case, it turns on the lysosome.
While the biology is complex, the idea is simple: increasing TFEB creates more lysosomal enzymes, more TPP1, more garbage collectors. It’s not a cure, but it could measurably slow the disease.
Sardiello has emerged as the James family’s primary therapeutic guide. They visited his lab in Houston last year, meeting with Sardiello and his boss, Huda Zoghbi.
“We agreed on a plan,” says Sardiello. “We would generate a mouse model that would recapitulate the same mutation found in Maya and test drugs against it. Some of those drugs might go to trials.”
To accelerate the process, Sardiello and his team are focused on approved drugs that have the potential to increase TFEB activity. Because these compounds have already been proven safe, the main hurdle will be showing efficacy.
A drug that boosts TFEB would be an incredible discovery for Maya and perhaps thousands of others. There are 50 lysosomal storage disorders that could potentially respond to a TFEB-based treatment, which might also benefit patients with Alzheimer’s, Parkinson’s, or Huntington’s.
“Lysosomal function is still involved because there’s an accumulation of aberrant proteins,” says Sardiello. “Our vision is that TFEB could be used to counteract the effects of many different diseases.”
Fighting for Maya
Setting up the collaboration with Baylor and the Sardiello lab was progress, but it didn’t come cheap. Beau and Suzette agreed to pay a postdoctoral researcher for four years—a $200,000 commitment.
The family has funded the first year, and supporters at Qualcomm and Maya’s school, San Diego Cooperative Charter (SDCCS), have embraced the cause. There have been jewelry sales, yoga lessons, and other fundraisers. People are making direct donations to the Sardiello lab, and every Friday both SDCCS campuses are flooded with #FightingForMaya T-shirts.

At home in Ocean Beach, the family has set up a stationary bike in their kitchen. Exercise can boost TPP1 levels. Maya understands that she must commit to her own treatment, but she’s also 13.
“She’ll ask how long she has to ride the bike, and we tell her six Meghan Trainor songs,” says Beau.
Their daily routine is filled with questions: Has she gone to the bathroom? Has she eaten enough? Does she have her water bottle?
Then there are the milestones: like a toddler’s, but reversed. In second grade, when Beau dropped her off at school, she’d run to the playground. In fifth grade she walked laps. Now she sits on a bench with her father and watches. On a good day she can tie her shoes.
“You see the progression,” says Suzette. “I can’t pack applesauce in a cup anymore because it’s too hard to get the lid off.”
Maya expresses herself as she can. A wave instead of hello. Her world is slowly contracting, but a new phone has helped. She relies on texting to relay family news, ask Beau how work went, or rat out her three younger siblings with a message like “Brothers mean.”
After this is over—and Sardiello or Pahan or BioMarin or Spark have come up with an effective treatment—Suzette and Beau will take a moment to breathe, and perhaps write a book so other families don’t have to start from scratch. But for now, they have to help Maya hold on to every tiny capability, because once they’re gone, they may never come back.
“I don’t have any other choice,” says Beau. “I have a great life, I work for a really amazing corporation, I solve complicated problems. I can do this for my family.”






Tags: Medical Research, Medicine, People